Nel capitolo precedente abbiamo parlato degli idrocarburi ciclici.
Prendiamo in considerazione il cicloesatriene.
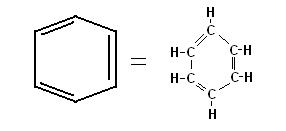
Osservando la struttura di questa molecola dovremmo aspettarci:
1. la possibilità di eseguire 3 addizioni elettrofile
2. una distanza tra gli atomi di carbonio interessati ai doppi legami diversa da quella esistente tra i carboni interessati ai legami singoli.
I chimici con diversi esperimenti hanno invece verificato che questa molecola:
1. non da reazioni di addizione, ma reazioni di sostituzione
2. le distanze tra tutti gli atomi di carbonio degli anelli sono uguali ed intermedie tra quelle di un doppio legame e di un legame singolo.
Questa discrepanza tra i dati previsti e quelli sperimentali osservati sulla molecola del cicloesatriene ha dato molto filo da torcere agli studiosi fino allo sviluppo della teoria della Risonanza.
Per comprendere questa teoria per prima cosa dobbiamo comprendere il significato di doppi legami coniugati.
Si parla di doppi legami coniugati quando in una molecola ci sono due doppi legami intervallati da un legame semplice.
Facciamo un esempio:
CH3-CH=CH–CH=CH-CH3 2,4 esadiene ha un doppio legame coniugato
CH3-CH=CH-CH2-CH=CH-CH3 2,5 eptadiene non ha un doppio legame coniugato
In un doppio legame coniugato gli elettroni non ibridi dei due doppi legami si trovano in una condizione particolare.
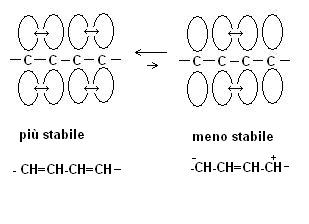
fig. 1
La vicinanza dei doppi legami consente infatti agli elettroni di poter essere “scambiati” anche dai due atomi di carbonio centrali.
Gli elettroni possono quindi “muoversi” su 4 atomi di carbonio anziché su due.
Questa situazione viene denominata delocalizzazione degli elettroni.
Nell’anello del cicloesatriene la delocalizzazione degli elettroni pigreco raggiunge una situazione “ottimale”.
Mentre le due forme della fig. 1 sono diversamente stabili ecco cosa succede nel cicloesatriene.
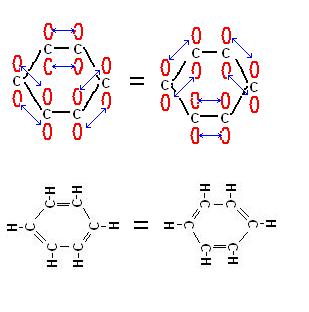
fig. 2
Le due forme possibili del ciclo esatriene sono del tutto analoghe.
Non essendoci differenza tra le due forme gli elettroni possono ritenersi completamente delocalizzati tra i 6 atomi di carbonio dell’anello.
Gli elettroni dei legami pigreco sono quindi in grado di ruotare liberamente sugli atomi di carbonio, dissipando in parte la loro energia.
La molecola diventa meno energetica e quindi più stabile.
Come abbiamo detto le due forme della figura 2 sono del tutto equivalenti tra di loro, esse però rappresentano solo delle condizioni “limite” denominate anche ibridi di risonanza.
Possiamo immaginare che nella realtà gli elettroni del cicloesatriene si trovino in una situazione intermedia tra quella dei due ibridi di risonanza.
I legami tra i carboni dell’anello si troveranno in una situazione intermedia tra quella di un legame sigma ed uno pigreco che contribuisce nel far assumere a questa molecola una struttura planare con gli elettroni pigreco che formano una sorta di nuvola elettronica sopra e sotto il piano.
Le caratteristiche che abbiamo illustrato spiegano bene le osservazioni sperimentali eseguite sul cicloesatriene (reazioni di sostituzione e lunghezza di legame).
La delocalizzazione degli elettroni tra due forme identiche prende anche il nome di risonanza ed i composti che possono dare questo fenomeno su anelli ciclici prendono il nome di idrocarburi aromatici.
Non possiamo quindi far ricadere il cicloesatriene nella categoria degli alcheni, ed a questo composto viene attribuito un nuovo nome: BENZENE.
Il benzene è il capostipite della famiglia degli idrocarburi aromatici è viene in genere rappresentato con il seguente simbolo:
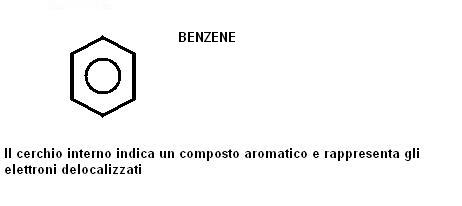
Il termine aromatico non è legato alle caratteristiche odorose di questi composti, ma dal fatto che sono stati estratti per la prima volta da piante aromatiche.
Dal benzene si originano diversi prodotti aromatici tra i quali possiamo citare:
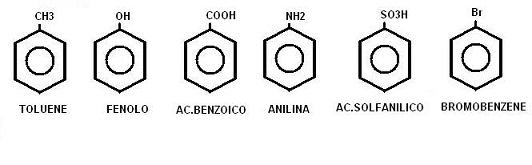
Per quanto riguarda la nomenclatura dei composti aromatici essa segue ancora molto spesso delle regole non IUPAC legate all’uso di nomi comuni.
Le posizioni sull’anello aromatico possono essere indicate numerandole da 1 a 6 partendo dal gruppo sostituente di maggiore importanza, oppure prendere il nome di posizione ORTO (equivalente ai numeri 2,6), posizione META (equivalente ai numeri 3,5) e posizione PARA (equivalente al numero 4).
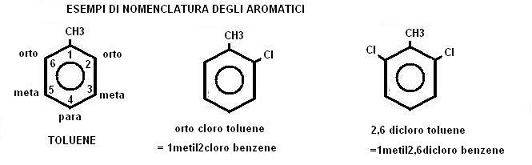
Caratteristiche chimico fisiche degli aromatici
Dal punto di vista delle proprietà fisiche, il benzene (ed i suoi derivati alchilici) non hanno sostanziali differenze con omologhi composti non aromatici a pari atomi di carbonio.
Si tratta per lo più di composti apolari, insolubili in acqua ed in genere liquidi a temperatura ambiente.
Per quanto riguarda le proprietà chimiche, esse possono risultare molto influenzate dalla natura dei gruppi sostituenti inseriti sull’anello aromatico, tuttavia in termini generali possiamo dire che le reazioni caratteristiche di questi composti sono le sostituzioni elettrofile.
Abbiamo già visto, parlando degli alcheni, che i reattivi elettrofili sono dal punto di vista chimico paragonabili agli acidi.
Si tratta di sostanze che sono avide di elettroni e come abbiamo visto nell’anello aromatico ci sono degli elettroni pigreco che possiamo ritenere appetibili.
Come mai allora i composti aromatici anziché dare reazioni di addizione elettrofila (come gli alcheni) danno reazioni di sostituzione elettrofila?.
La risposta è legata ad un discorso di stabilità.
Abbiamo visto come i composti aromatici siano dei composti stabili, in quanto la delocalizzazione degli elettroni “produce” stabilità.
Perché la risonanza sia presente su di un anello occorre che ci siano tre doppi legami coniugati.
Se avvenisse una reazione di addizione elettrofila su di un composto aromatico, si verrebbe a perdere un doppio legame e quindi la possibilità di risonanza.
In altre parole da una addizione elettrofila si otterrebbe un prodotto meno stabile di quello di partenza.
Le reazioni di sostituzione non modificano la natura dei legami (per questo in genere interessano composti stabili) e quindi consentono al prodotto di mantenere le caratteristiche aromatiche.
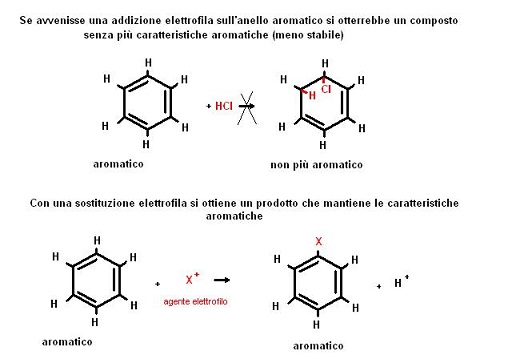
Vediamo allora quali sono le principali reazioni di sostituzione elettrofila sui composti aromatici e quali reagenti prevedono:
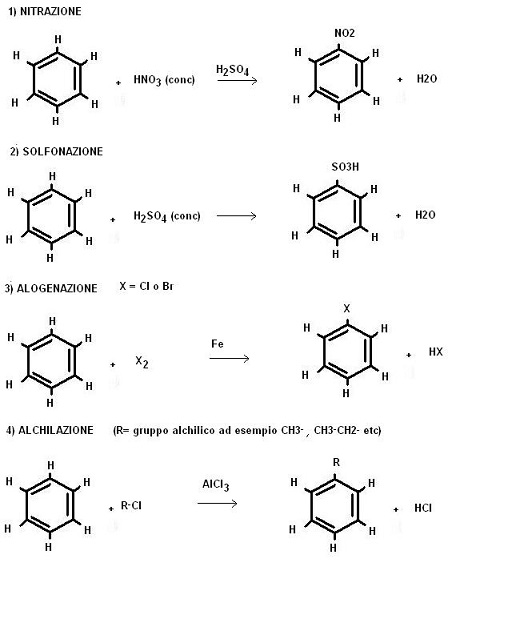
Il meccanismo di queste reazioni è analogo per tutte le casistiche, immaginando un generico agente elettrofilo XZ (X+= agente elettrofilo, Z–= agente nucleofilo) possiamo così schematizzare:
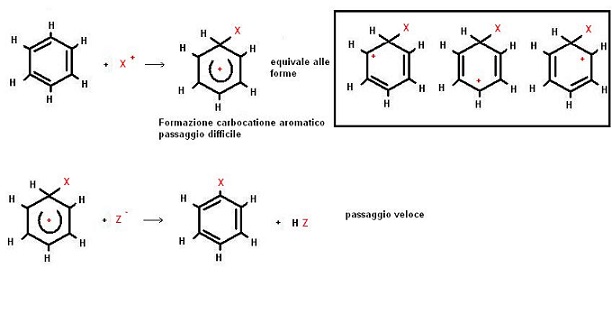
Anche in questo caso il passaggio lento corrisponde alla formazione del carbocatione.
Una delle conseguenze di tale fatto consiste nell’orientamento seguito dalle reazioni di sostituzione elettrofila in composti aromatici già sostituiti.
Se prendiamo ad esempio il toluene e proviamo a fare una nitrazione otterremo come composti principali l’orto ed il para nitro toluene, mentre partendo dal fenolo otterremo il meta nitro fenolo. A cosa è dovuta questa differenza.
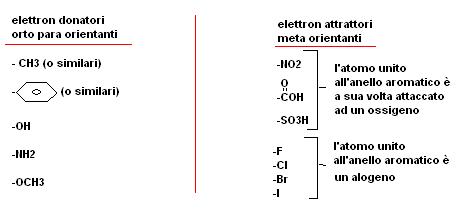
In pratica i gruppi sostituenti presenti sull’anello aromatico si possono dividere in due grandi categorie:
· gli orto, para orientanti
· i meta orientanti.
Con una certa semplificazione possiamo dire che i gruppi che hanno caratteristiche elettron attrattrici sono meta orientanti, mentre i gruppi che risultano elettron donatori sono orto-para orientanti.
Per distinguere un gruppo elettron donatore da un gruppo elettron attrattore possiamo fare la seguente considerazione:
· se l’atomo attaccato all’anello è a sua volta unito ad un atomo “poco” elettronegativo il gruppo è elettron donatore
· se l’atomo attaccato all’anello è a sua volta unito ad un atomo “molto” elettron donatore (in genere ossigeno o azoto) il gruppo è elettron attrattore.
Nella seguente tabella sono riportati i principali gruppi elettron attrattori ed elettron donatori
Se si esegue una sostituzione elettrofila su un composto aromatico in cui è già presente un gruppo sostituente, si possono formare tre terne di carbocationi (tre per l’attacco in posizione orto, tre per l’attacco in posizione meta e tre per l’attacco in posizione para).
Tra questi carbocationi sono due presentano il “+” attaccato al gruppo sostituente già presente.
Come si può osservare dalla figura sotto riportata le due forme di carbocatione evidenziate in giallo in cui il “+” è unito al carbonio già sostituito sono relative all’attacco orto e para.
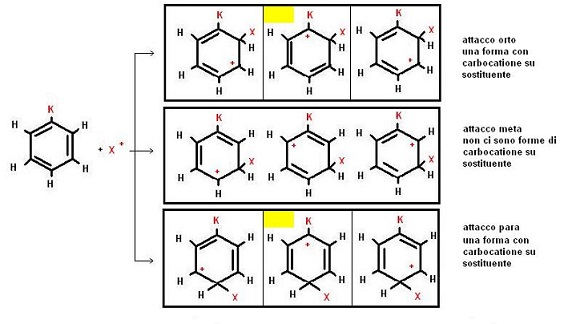
In pratica se K è elettron donatore le forme evidenziate in giallo saranno favorite, e quindi risulterà più probabile l’attacco orto, para.
Se, invece, K è elettron attrattore le forme evidenziate in giallo saranno sfavorite, e quindi risulterà più probabile l’attacco in posizione meta.